




Ukierunkowany opatrunek na amplikon i sekwencjonowanie w nanoskali
Zaprojektowano zestaw zestawów starterów, protokołów i poleceń obliczeniowych do sekwencjonowania genomów hMPXV1 w komórce przepływowej nanoporów MinION (ryc. 2a). Wszystkie niezbędne protokoły zostały pokrótce opisane w sekcji Metody i są dostępne jako plik uzupełniający 2: Protokoły laboratorium roboczego. Pierwotne startery do amplikonów zaprojektowano przy użyciu PrimalScheme [9], który konstruuje sekwencję kafelkowych amplikonów wielkości klastra przy użyciu znanego szablonu genetycznego, przeciwko sekwencji referencyjnej NCBI NC_063383.1, sekwencji Clade IIb, Lineage A z 2018 r. i niedawnym próbkom epidemii, dopasował sześć różnych genomów obejmujących próbki ospy od 2003 do współczesność Zaprojektowane przedrostki zostały z nimi porównane. Zachowano tylko startery pasujące do sześciu genomów, co wskazuje, że zamieszkiwały one regiony o niskiej zmienności genetycznej. Te, które zawierają niezgodności, zostały przeprojektowane. Zapewniają one najwyższe prawdopodobieństwo, że startery zadziałają z nowymi mutacjami choroby ospy podczas tej epidemii iw przyszłości. Ostateczny projekt obejmował 73 kafelkowe amplikony o długości ~3000 pz, które podzielono na dwie grupy. Pula 1 zawierała startery dla amplikonów o numerach nieparzystych, a pula 2 zawierała startery dla amplikonów o numerach parzystych (ryc. 2b). Ze względu na regiony powtórzeń końcowych (~ 6400 pz) na końcach genomu, znane jako odwrócone powtórzenia końcowe (ITR), amplikony 1 i 73 mają te same startery, co amplikony 2 i 72. Te same nakładające się startery zewnętrzne, ale różne wewnętrzne podkłady .
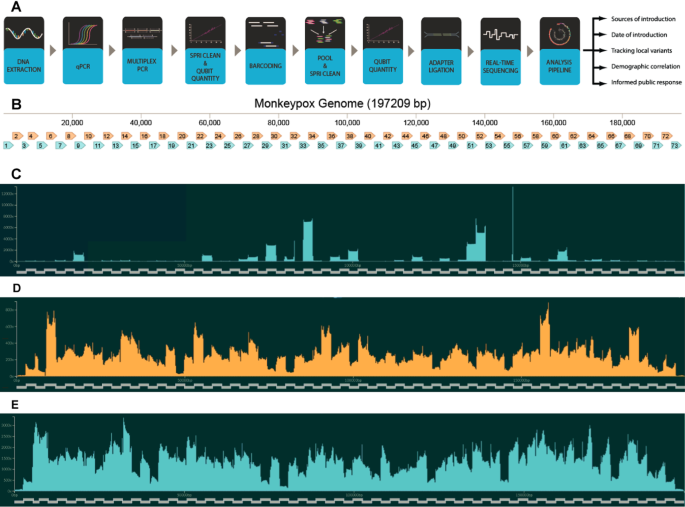
A Diagram ilustrujący przebieg procesu przygotowywania i sekwencjonowania próbki pobranej od ospy. B Mapa amplikonu genomu Mpox. Amplikony zespołu 1 są pokazane na niebiesko, a amplikony zespołu 2 są pokazane na pomarańczowo. Amplikony 1 i 73 pasują do siebie nawzajem, podobnie jak amplikony 2 i 72. CE-E Pokrycie poszczególnych amplikonów w genomie Mpox podczas sekwencjonowania. FASTQ pochodzi z odczytów przy użyciu programu RAMPART [7]. Amplikony kafelkowe są pokazane jako oś x. Oś Y wskazuje liczbę odczytów pasujących do tego locus w genomie. C Pierwsza próba sekwencjonowania genomu USA-2003 z wykorzystaniem USA-2003 jako genomu referencyjnego. (dr) podjęli próbę udoskonalonego sekwencjonowania z USA-2003. H Przykład próby sekwencjonowania genomu niedawnej epidemii z 2022 r., przy użyciu sekwencji linii B.1 jako genomu odniesienia
Amplifikację PCR testowano stosując pule starterów przeciwko genomowi znanego wirusa ospy wietrznej USA-2003 z wybuchu epidemii w Wisconsin w 2003 r., hodowanego w komórkach zielonej małpy (BEI Resources, NR-4928). Ponieważ próbki handlowe zawierały niską liczbę kopii wirusa, przed amplifikacją PCR po raz pierwszy zastosowano bezstronną metodę amplifikacji całego genomu. Otrzymane amplikony z każdej puli starterów połączono po PCR, oznaczono kodami kreskowymi i zsekwencjonowano na narzędziu MinION. Odczyty wykorzystano do wygenerowania sekwencjonowania konsensusu. Ta wstępna próba PCR i sekwencjonowania dała 20-krotne pokrycie 78,3%, co ogranicza pozyskiwanie całego genomu, ponieważ narzędzia takie jak Nanopolish wymagają 20-krotnego pokrycia, aby wywołać sekwencję konsensusową. Dystrybucja amplikonów była przesunięta w kierunku produkcji dużej liczby z garstki amplikonów. Ponadto tylko 52% wszystkich odczytów zmapowano na genom ospy, co wskazuje na niespecyficzną amplifikację (ryc. 2c).
Po dokładnej analizie zidentyfikowano i zastąpiono startery poddane amplifikacji poza celem amplifikacji genomowego DNA zielonej małpy lub ludzkiego. Stwierdziliśmy również, że amplikony 18, 20, 38, 40, 43 i 52 były nieco niedoreprezentowane, podczas gdy amplikony 32 i 51 były nadreprezentowane. Następnie podwoiliśmy stężenie starterów dla amplikonów 18, 20, 38, 40, 43 i 52. Inne zmiany obejmowały ulepszone warunki cykli termicznych i zmianę kodowania kreskowego z metody opartej na splicingu na szybką macierz kodów kreskowych opartą na transpozazie, która dała lepsze i szybsze rezultaty. . Powtórzona próba sekwencjonowania z wykorzystaniem tych zmian miała 20-krotny zasięg 97,10% i 97,42% odczytów dopasowanych do genomu USA-2003, co wskazuje na czystą amplifikację PCR (ryc. 2D). Stwierdzono, że uzyskana sekwencja konsensusowa dokładnie pasuje do przewidywanego genomu USA-2003 we wszystkich regionach o wystarczającym pokryciu.
Następnie opublikowaliśmy tę metodę sekwencjonowania próbek obecnych pacjentów z ospą prawdziwą w 2022 roku z Illinois w ciągu pierwszych kilku miesięcy wybuchu epidemii. Przeprowadzono ekstrakcję kwasów nukleinowych na bazie kulek magnetycznych, a następnie oszacowano miano wirusa za pomocą ilościowego PCR (qPCR). Obiekty badawcze, w których pobrano próbki, wykorzystywały ogólny materiał inny niż wapienne ortopokswirus (NVO) sonda qPCR zaprojektowana przez Centrum Zapobiegania i Kontroli Chorób (CDC) do badania obecności DNA wirusa ospy. Inne placówki badawcze mogą stosować qPCR w oparciu o sondę CDC dotyczącą ospy. Chociaż ta sonda jest dokładniejsza w wykrywaniu ospy, zawiodła w niewielkim podzbiorze próbek ospy, które po raz pierwszy zaobserwowano w Kalifornii. Wynika to z częściowych delecji ITR, w których wiąże się sonda qPCR. Próbki te zidentyfikowano wyłącznie za pomocą testu NVO qPCR. Aby zabezpieczyć się przed podobnymi problemami, zaprojektowaliśmy opartą na sondzie reakcję qPCR, OPG123, opartą na analizie regionów o niskiej mutacji w genomie mpox. Aby ustandaryzować odczyty, próbkę DNA przetestowano w tych samych warunkach qPCR i stosując tę samą wyjściową wartość progową. Ogólnie rzecz biorąc, nasz próg cyklu (CR) wartości wynosiły około czterech CR wyższe wartości niż te generowane przez obiekty badawcze (dodatkowy plik 1: rysunek S1). Może to być spowodowane różnymi warunkami qPCR, degradacją podczas transportu lub cyklami zamrażania i rozmrażania.
Aby określić liczbę genomów potrzebnych do pomyślnego sekwencjonowania, zsyntetyzowano krótki fragment dwuniciowego DNA i zastosowano go do amplifikacji ze znaną liczbą kopii (plik uzupełniający 1: rysunek S2). Na podstawie tych wyników potrzeba od 5000 do 10 000 kopii genomu Mpox, aby wyprodukować optymalny amplikon i wystarczające pokrycie do sekwencjonowania genomu. Jest to związane z CR Wartość 32 lub mniej z naszą sondą qPCR OPG123. Jednak w oparciu o porównania qPCR między naszą sondą, sondą CDC specyficzną dla ospy i sondą RNazy P (ludzki genomowy DNA), ta redukcja liczby kopii może być powiązana z ilością DNA ospy prawdziwej w porównaniu z zanieczyszczeniem ludzkiego DNA (Uzupełniający plik 1: Rysunek S3). Próbki z niskim OPG123 CR Wartości mają również wysoki stosunek DNA Mpox do ludzkiego DNA (8-12 CR różnica), podczas gdy próbki z wysokim OPG123 CR Wartości mają niższe stosunki DNA Mpox do ludzkiego DNA (3-4 CR różnica).
Po oznaczeniu ilościowym qPCR genomowy DNA hMPXV1 amplifikowano oddzielnie starterem z puli 1 i puli 2. Otrzymane amplikony z dwóch reakcji następnie połączono i poddano serii etapów oczyszczania i przygotowania biblioteki (patrz plik uzupełniający 2: protokoły pracy). Po sekwencjonowaniu nanoporów, które można osiągnąć przez zakodowanie do 24 próbek na pojedynczej komorze przepływowej, dla każdej próbki wygenerowano sekwencję konsensusową. W sumie przetworzono 103 próbki pacjentów, uzyskując 84 całe genomy w oparciu o sekwencjonowanie konsensusowe. Dodatkowy plik 1: Pokazana jest tabela S1 CR wartości i liczby pokrycia 100x/20x dla tych próbek 84. Średnie pokrycie sekwencji konsensusu wyniosło 98,79%, przy średnim pokryciu 99,41%. Przykład 99,95% pokrycia sekwencji, jak pokazano przez Rampart, pokazano na ryc. 2E. Jednym z wyzwań związanych z pokryciem genomu była ostateczna replikacja ITR. Minimap2, oprogramowanie używane do wyrównywania odczytów z szablonem genomu, próbuje dopasować te odczyty do jednego lub drugiego końca genomu, ale nie do obu. Jest to problem, jeśli w tym obszarze jest tylko kilka odczytów. Może to prowadzić do nieuzasadnionych zasad (Ns) w amplikonach 1 i 73 w ostatecznym konsensusie. Jeśli generowane są niewystarczające odczyty, jedną ze strategii jest usunięcie regionu powtórzeń końcowych z jednego końca genomu matrycy, tak aby minimapa2 wyrównała wszystkie odczyty na drugim powtórzeniu końcowym. Usunięty duplikat można następnie ręcznie dodać ponownie.
Różnorodne wprowadzenia wirusowe
Po sekwencjonowaniu wszystkie sekwencje konsensusowe analizowano przy użyciu oprogramowania NextClade [10]. Porównuje to sekwencję będącą przedmiotem zainteresowania z genomem generycznego modelu Clade IIb, linii B.1, opartego na ON563414.3, MPXV_USA_2022_MA001, próbce z obecnego wybuchu epidemii zebranej w maju 2022 r. W Massachusetts, USA, i podkreśla wszelkie indele lub mutacje . Sekwencje można następnie podzielić na określone podklady w obrębie B1. Od października 2022 r. Istnieje dwanaście podkladów, zgodnie z definicją grupy NextClade [11] (Rys. 1b). Każdy ma od jednej do czterech unikalnych mutacji nukleotydów. Istnieje również około 60% sekwencji obecnej epidemii, które należą do szczepu B.1, ale nie pasują do określonego podgatunku. Nasza sekwencja wskazuje, że Illinois doświadczyło dużej liczby różnych introdukcji wirusowych. Tabela 1 pokazuje niektóre mutacje, które napotkano w naszym sekwencjonowaniu.
Ponadto osiem innych sekwencji posiada unikalne mutacje, które nie są wspólne z żadną inną sekwencją w bazie danych NCBI. Te różnorodne wzorce mutacji w niewielkiej liczbie przypadków wskazują, że duża liczba wersji wirusowych weszła do regionu i rozprzestrzeniła się.
Odkryj nowy rodowód B 1
Sekwencja genomu wskazuje również, że kilka szczepów B.1 mogło pojawić się w regionie Illinois lub Midwest. Spośród 84 zsekwencjonowanych próbek jedna czwarta miała cztery wspólne mutacje: C18133T, G67611A, G130231A i G159277A (ryc. 3). Trzy z tych mutacji są zlokalizowane w regionach niekodujących, podczas gdy ostatnia, G159277A, skutkuje mutacją E121K w białku OPG185. W czasie tego sekwencjonowania ta klasyfikacja mutacji była nieokreślona. Został później sklasyfikowany jako Lineage B.1.11 przez Nextclade. Obecnie B.1.11 jest piątą najpowszechniejszą linią w obecnej epidemii Mpox, z łącznie osiemdziesięcioma czterema sekwencjami. Nasze sekwencje stanowiły 25% genomów potrzebnych do ustanowienia tej nowo nazwanej linii. Linia ta występuje głównie w Stanach Zjednoczonych, z zaledwie kilkoma próbkami spoza Ameryki Północnej: infekcje sierpniowe i wrześniowe z Kolumbii, infekcje z początku sierpnia w Portugalii i infekcje wrześniowe w Niemczech. Chociaż niedostatek sekwencji utrudnia dokładne poznanie pochodzenia tej linii, wszystkie pierwsze przypadki wykryto w Illinois pod koniec czerwca. Podobne szczepy pojawiły się w Kalifornii, Waszyngtonie i innych częściach Stanów Zjednoczonych dopiero w lipcu
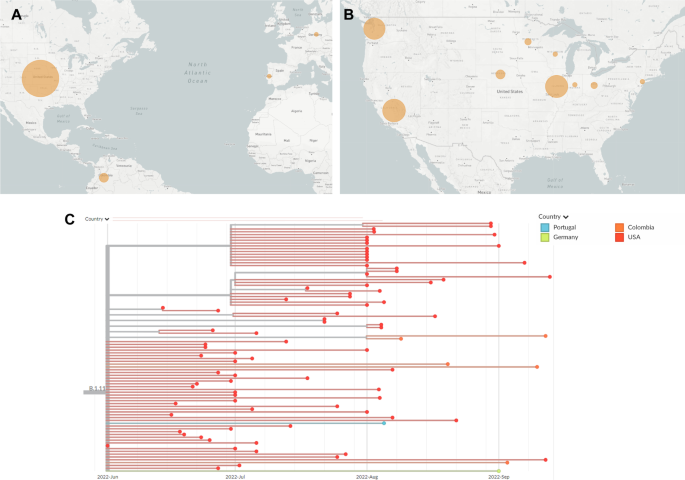
Dane geograficzne i filogenetyczne dla linii Mpox B 1.11. A Mapa świata przedstawiająca rozmieszczenie przodków według krajów. Im większe koło, tym więcej sekwencji. Porównaj z całkowitymi sekwencjami na ryc. 1c. B Mapa Stanów Zjednoczonych przedstawiająca rozmieszczenie przodków według stanów. Porównaj z całkowitymi sekwencjami na ryc. 1d. C Drzewo filogenetyczne przedstawiające daty i lokalizacje zbioru dla każdej sekwencji z B.1.11
Inny główny wzorzec mutacji obserwowany w naszych sekwencjach dopiero niedawno otrzymał przypisanie linii, B.1.13. (Rys. 4). Dziewiętnaście z naszych osiemdziesięciu czterech próbek miało G175093A, a osiemnaście miało wtórną mutację C132520T. G175093A nadaje mutację D188N w białku OPG204, podczas gdy C132520T znajduje się w regionie niekodującym. B.1.13 występuje przede wszystkim w środkowo-zachodnich Stanach Zjednoczonych i wydaje się, że pochodzi stamtąd, a infekcja tymi mutacjami pojawiła się w połowie czerwca. Stamtąd wydaje się, że stopniowo rozprzestrzenił się na inne obszary Środkowego Zachodu, takie jak Oklahoma, Minnesota, Ohio, Tennessee i Kentucky. Do tej pory żadne sekwencje z tą mutacją nie pojawiły się w Kalifornii, a tylko kilka w stanie Waszyngton, pomimo wysokiego wskaźnika sekwencjonowania. Wreszcie, pod koniec lipca podobną sekwencję zaobserwowano w Wielkiej Brytanii, a pod koniec sierpnia w grupie w Niemczech zaobserwowano inną sekwencję.
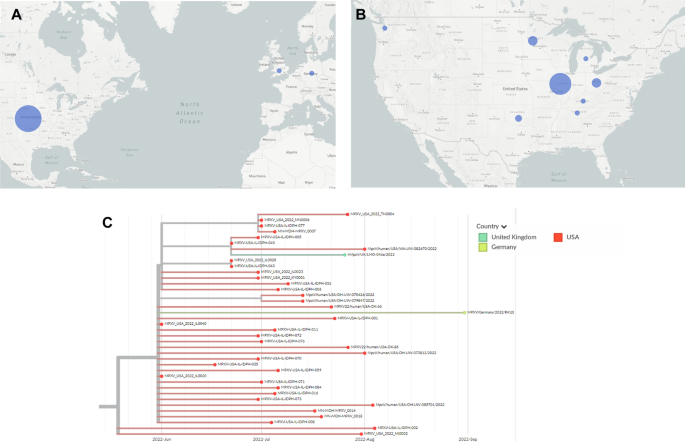
Dane geograficzne i filogenetyczne dla linii Mpox B 1.13. A Mapa świata przedstawiająca rozmieszczenie przodków według krajów. Im większe koło, tym więcej sekwencji. Porównaj z całkowitymi sekwencjami na ryc. 1c. B Mapa Stanów Zjednoczonych przedstawiająca rozmieszczenie przodków według stanów. Porównaj z całkowitymi sekwencjami na ryc. 1d. C Drzewo filogenetyczne przedstawiające daty i lokalizacje zbioru dla każdej sekwencji w b 1.13
Obecnie B.1.13 jest jedenastym najczęściej występującym szczepem podczas globalnej epidemii w 2022 r., z łącznie trzydziestoma siedmioma sekwencjami. Połowa sekwencji została wygenerowana w b 1.13 tego badania. Ilustruje to potrzebę głębszego sekwencjonowania regionalnego i zdolność niewielkiej liczby regionalnych genomów do wpływania na globalną epidemiologię.

Dorota Masłowska jest współpracowniczką serwisu Przecław News, gdzie pisze o wiadomościach, polityce, biznesie, technologii, sporcie, rozrywce i stylu życia. Koncentruje się na dostarczaniu przejrzystych i użytecznych informacji o aktualnych wydarzeniach, pomagając czytelnikom śledzić najważniejsze tematy oraz historie istotne dla codziennego życia.




Więcej artykułów
Pragmata – ambitny projekt Capcomu w czasach kryzysu branży
Załoga Artemis II obserwuje niewidoczną stronę Księżyca. Przełomowa misja NASA
Prognoza cukrzycy w Australii w 2024 r. | Wiadomości o Mirażu